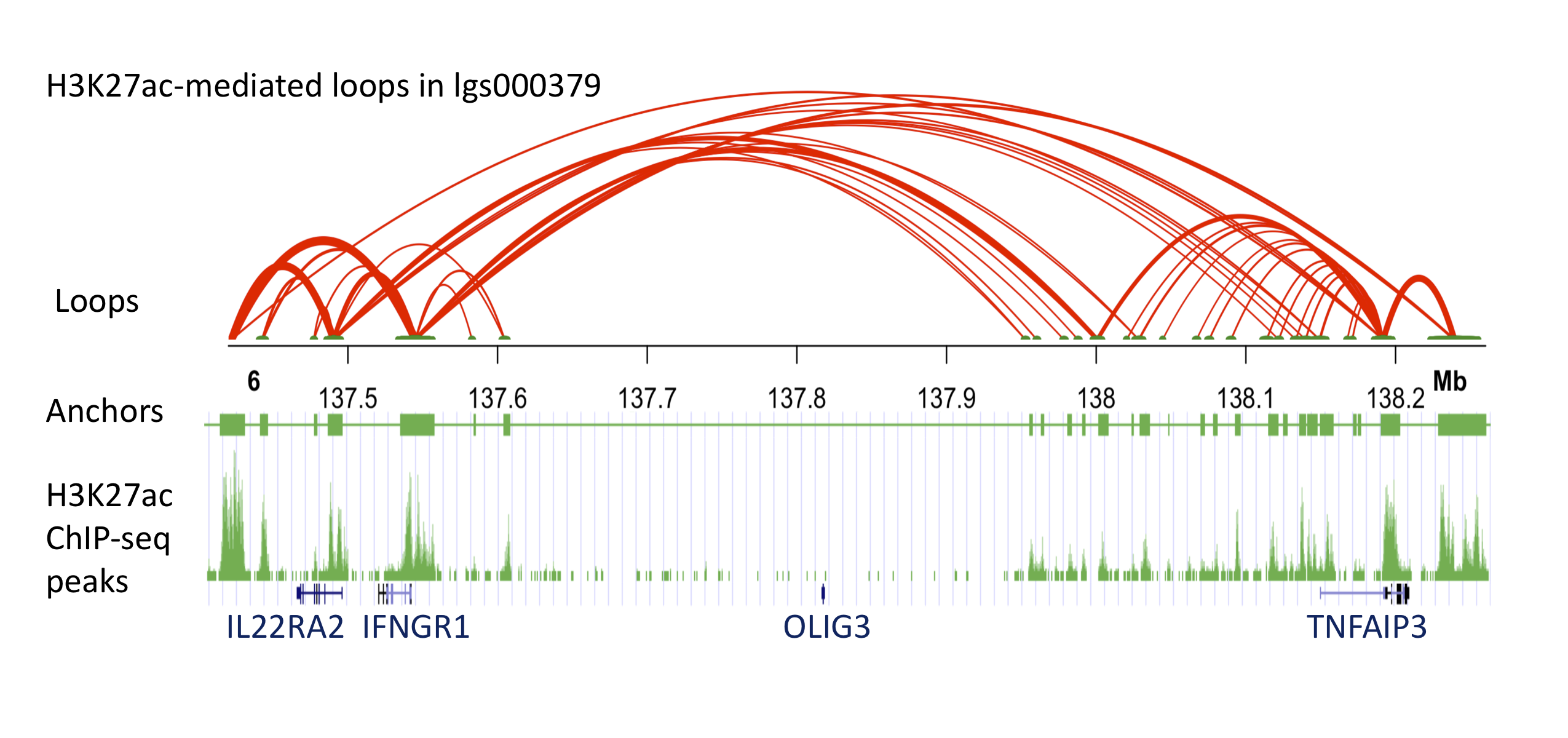
 Three-dimensional (3D) chromatin loops are critical for transcriptional regulation by bringing distant promoters and enhancers into close proximity. Protein factors, such as cohesin and CCCTC-binding factor (CTCF) facilitate the formation of the functional 3D chromatin conformation. Therefore, studying the protein-mediated 3D genome architecture can help to understand how specific genes contribute to human diseases. HiChIP is a new method to identify protein-mediated 3D chromatin looping information, and it was shown to have higher efficiency and specificity compared to Hi-C for the purpose of identifying such loops. In this study, we performed HiChIP assays to map the DNA looping patterns in EBV-transformed human B cell lines originally obtained from 3 systemic lupus erythematosus (SLE) patients. We measured 3D DNA contacts mediated by an essential nucleus protein, CTCF, and an epigenetic histone mark, histone 3 lysine 27 acetylation (H3K27ac). We also performed ChIP-seq assays to refine accurate anchor ranges. Using a minimum input of 1 million cells, we obtained approximately 100M paired-end tags (PETs), of which 30% represent intrachromosomal long-range interactions spanning between 5KB and 2MB. Distinct anchor and looping patterns were observed when targeting the two nucleus markers. The H3K27ac HiChIP analyses showed more cell-specific loops in enhancer-promoter regions, compared to CTCF HiChIPs, supporting its known role as an active enhancer marker throughout the genome. We further investigated the chromatin interactions approximately 185kb upstream of the tumor necrosis factor alpha inducible protein 3 (TNFAIP3) gene. This non-coding region was previously identified as an SLE and rheumatoid arthritis (RA) susceptibility region, however, the function of associated SNPs (e.g. rs10499194, rs6920220) in this region is still not clear. Our H3K27ac HiChIP data showed that this region is enriched with H3K27ac-mediated looping activities. Interestingly, although this region is flanked by the oligodendrocyte transcription factor 3 (OLIG3) and TNFAIP3 gene, we found that the associated SNPs are mainly looping to further upstream genes, interleukin 22 receptor alpha 2 (IL22RA2) and interferon gamma receptor 1 (IFNGR1). Our data provide new insight into finding direct functional targets associated with GWAS variants that are in non-coding regions. The data can also help to identify potential causal risk SNPs within a large linkage disequilibrium (LD) region.
Three-dimensional (3D) chromatin loops are critical for transcriptional regulation by bringing distant promoters and enhancers into close proximity. Protein factors, such as cohesin and CCCTC-binding factor (CTCF) facilitate the formation of the functional 3D chromatin conformation. Therefore, studying the protein-mediated 3D genome architecture can help to understand how specific genes contribute to human diseases. HiChIP is a new method to identify protein-mediated 3D chromatin looping information, and it was shown to have higher efficiency and specificity compared to Hi-C for the purpose of identifying such loops. In this study, we performed HiChIP assays to map the DNA looping patterns in EBV-transformed human B cell lines originally obtained from 3 systemic lupus erythematosus (SLE) patients. We measured 3D DNA contacts mediated by an essential nucleus protein, CTCF, and an epigenetic histone mark, histone 3 lysine 27 acetylation (H3K27ac). We also performed ChIP-seq assays to refine accurate anchor ranges. Using a minimum input of 1 million cells, we obtained approximately 100M paired-end tags (PETs), of which 30% represent intrachromosomal long-range interactions spanning between 5KB and 2MB. Distinct anchor and looping patterns were observed when targeting the two nucleus markers. The H3K27ac HiChIP analyses showed more cell-specific loops in enhancer-promoter regions, compared to CTCF HiChIPs, supporting its known role as an active enhancer marker throughout the genome. We further investigated the chromatin interactions approximately 185kb upstream of the tumor necrosis factor alpha inducible protein 3 (TNFAIP3) gene. This non-coding region was previously identified as an SLE and rheumatoid arthritis (RA) susceptibility region, however, the function of associated SNPs (e.g. rs10499194, rs6920220) in this region is still not clear. Our H3K27ac HiChIP data showed that this region is enriched with H3K27ac-mediated looping activities. Interestingly, although this region is flanked by the oligodendrocyte transcription factor 3 (OLIG3) and TNFAIP3 gene, we found that the associated SNPs are mainly looping to further upstream genes, interleukin 22 receptor alpha 2 (IL22RA2) and interferon gamma receptor 1 (IFNGR1). Our data provide new insight into finding direct functional targets associated with GWAS variants that are in non-coding regions. The data can also help to identify potential causal risk SNPs within a large linkage disequilibrium (LD) region.