 The human gut is home to tens of trillions of microorganisms (bacteria, viruses, yeasts, fungi, and protozoans) that function in symbiosis with host cells to support digestion, metabolism, and immunity. Specifically, gut microbiota educates host immune cells to protect against invading pathogens and loss of self-tolerance. Identifying and characterizing the functional mechanisms and regulatory machinery by which the gut microbiome influences health and disease are ongoing initiatives of the NIH Human Microbiome Project. Gut dysbiosis – disease-associated disruptions or alterations in the gut microbiota diversity – occurs in cancers, cardiovascular disease, neurological dysfunction, and autoimmune diseases. Whether gut dysbiosis is a cause or consequence of pathogenesis is largely debated.
The human gut is home to tens of trillions of microorganisms (bacteria, viruses, yeasts, fungi, and protozoans) that function in symbiosis with host cells to support digestion, metabolism, and immunity. Specifically, gut microbiota educates host immune cells to protect against invading pathogens and loss of self-tolerance. Identifying and characterizing the functional mechanisms and regulatory machinery by which the gut microbiome influences health and disease are ongoing initiatives of the NIH Human Microbiome Project. Gut dysbiosis – disease-associated disruptions or alterations in the gut microbiota diversity – occurs in cancers, cardiovascular disease, neurological dysfunction, and autoimmune diseases. Whether gut dysbiosis is a cause or consequence of pathogenesis is largely debated.
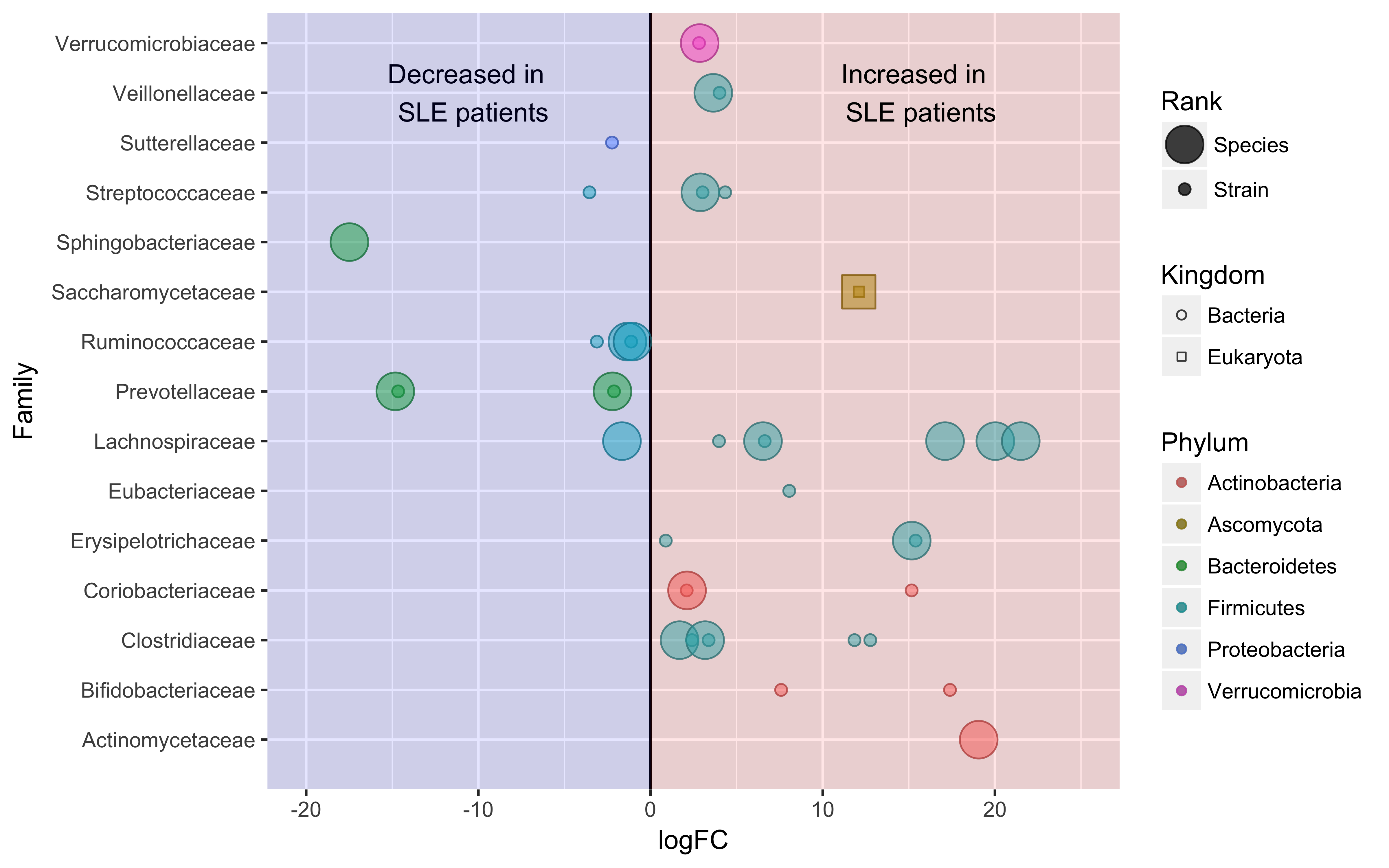
Systemic lupus erythematosus (SLE) is a heritable and progressive autoimmune disease of unknown etiology. SLE is characterized by a loss of tolerance against a wide variety of self-antigens and dysregulated interferon responses that cause chronic inflammation and, ultimately, end-organ damage to multiple organs. Clinical heterogeneity between afflicted family members with SLE is indicative of a strong environmental component that influences genetic penetrance of SLE-associated risk genes and promotes the production of autoantibodies. Gut dysbiosis of SLE patients has been implicated as an important environmental factor contributing to SLE pathogenesis. However, the taxonomical and functional characterization of gut dysbiosis in SLE remains largely underexplored.
In this study, we performed 16S survey to broadly characterize SLE-associated microbiota, then followed-up with Whole Metagenome Shotgun (WMS) sequencing to obtain deeper taxonomic profiles and directly characterize the differences in functional potential between healthy and SLE-associated microbiota. In summary, the diversity metrics gathered using two separate experimental approaches suggest that the gut microbiota of SLE patients is not correlated with broad, whole-community structural changes, but rather differs at deeper taxonomical ranks, i.e. species and strains, and in the functional capabilities associated with the microbiome.
To our knowledge, this is the first analysis of SLE-associated microbiota using WMS sequencing. Our findings suggest that SLE patients do indeed have gut dysbiosis, and that the species and strains identified as altered in SLE samples may exacerbate systemic inflammatory responses thought to exacerbate SLE pathogenesis. Further research in this domain may be required to isolate and characterize experimentally identified species, strains or viral factors, and the key metabolites derived from them. We hope to guide cultivation efforts to investigate these members beyond statistical associations to understand the underpinning mechanisms relating to SLE pathogenesis and drive further research towards functional testing on animal models.