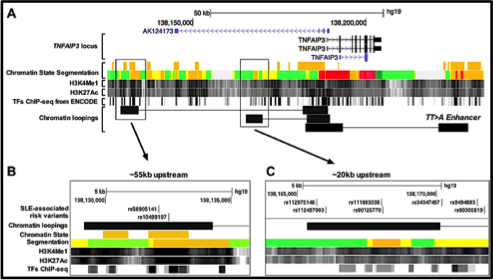
 A key objective of human genetics is to identify and characterize variants associated with complex diseases. In this study, we propose that a regulatory variant (rs10499197) situated 55kb upstream of TNFAIP3 is a likely candidate associated with SLE. TNFAIP3 encodes A20, an ubiquitin-editing enzyme that inhibits Nuclear Factor-kappa B (NF-κB) signaling to modulate innate and adaptive immune responses. Loss of TNFAIP3 expression is implicated in the pathogenesis of autoimmune disease; however, mechanisms responsible for impaired expression remain unclear. We used a combination of electrophoretic mobility shift assays (EMSA), reporter assays, chromatin immunoprecipitation-PCR (ChIP-PCR), Hi-ChIP, and chromosome conformation capture (3C) assays on EBV-transformed B cell lines from individuals carrying risk and non-risk TNFAIP3 haplotypes to characterize the effect of rs10499197 on TNFAIP3 expression. Our results show that rs10499197 resides in an enhancer element enriched with transcription factor binding sites. Also, we show that this upstream regulatory element physical interacts with the TNFAIP3 promoter through long-range DNA looping. Presence of the risk allele inhibits the looping interaction resulting in reduced A20 expression. Results suggest that the SLE risk variant rs10499197 is potentially a causal variant that disrupts the function of a putative enhancer upstream of TNFAIP3. Disrupting the enhancer impairs TNFAIP3 expression, enhances NF-κB signaling, and heightens immune responses in a cell type-specific manner. Following our work on the TT>A enhancer, this is the second likely causal variant to impact TNFAIP3 expression.
A key objective of human genetics is to identify and characterize variants associated with complex diseases. In this study, we propose that a regulatory variant (rs10499197) situated 55kb upstream of TNFAIP3 is a likely candidate associated with SLE. TNFAIP3 encodes A20, an ubiquitin-editing enzyme that inhibits Nuclear Factor-kappa B (NF-κB) signaling to modulate innate and adaptive immune responses. Loss of TNFAIP3 expression is implicated in the pathogenesis of autoimmune disease; however, mechanisms responsible for impaired expression remain unclear. We used a combination of electrophoretic mobility shift assays (EMSA), reporter assays, chromatin immunoprecipitation-PCR (ChIP-PCR), Hi-ChIP, and chromosome conformation capture (3C) assays on EBV-transformed B cell lines from individuals carrying risk and non-risk TNFAIP3 haplotypes to characterize the effect of rs10499197 on TNFAIP3 expression. Our results show that rs10499197 resides in an enhancer element enriched with transcription factor binding sites. Also, we show that this upstream regulatory element physical interacts with the TNFAIP3 promoter through long-range DNA looping. Presence of the risk allele inhibits the looping interaction resulting in reduced A20 expression. Results suggest that the SLE risk variant rs10499197 is potentially a causal variant that disrupts the function of a putative enhancer upstream of TNFAIP3. Disrupting the enhancer impairs TNFAIP3 expression, enhances NF-κB signaling, and heightens immune responses in a cell type-specific manner. Following our work on the TT>A enhancer, this is the second likely causal variant to impact TNFAIP3 expression.