 SLE is a prototype systemic autoimmune disease. Genome-wide association studies have fine mapped a 67kb risk haplotype that spans the Ubiquitin-conjugating enzyme E2 L3 (UBE2L3) gene to confer disease risk in SLE. UBE2L3 encodes an E2 ubiquitin-conjugating enzyme (UbcH7) involved in the ubiquitin signaling pathway. The UBE2L3 risk haplotype is found to significantly increase UBE2L3 expression. In this study, we aim to identify the potential SLE causal variant candidates among the single nucleotide polymorphisms spanning UBE2L3 risk haplotype that can increase UBE2L3 expression.
SLE is a prototype systemic autoimmune disease. Genome-wide association studies have fine mapped a 67kb risk haplotype that spans the Ubiquitin-conjugating enzyme E2 L3 (UBE2L3) gene to confer disease risk in SLE. UBE2L3 encodes an E2 ubiquitin-conjugating enzyme (UbcH7) involved in the ubiquitin signaling pathway. The UBE2L3 risk haplotype is found to significantly increase UBE2L3 expression. In this study, we aim to identify the potential SLE causal variant candidates among the single nucleotide polymorphisms spanning UBE2L3 risk haplotype that can increase UBE2L3 expression.
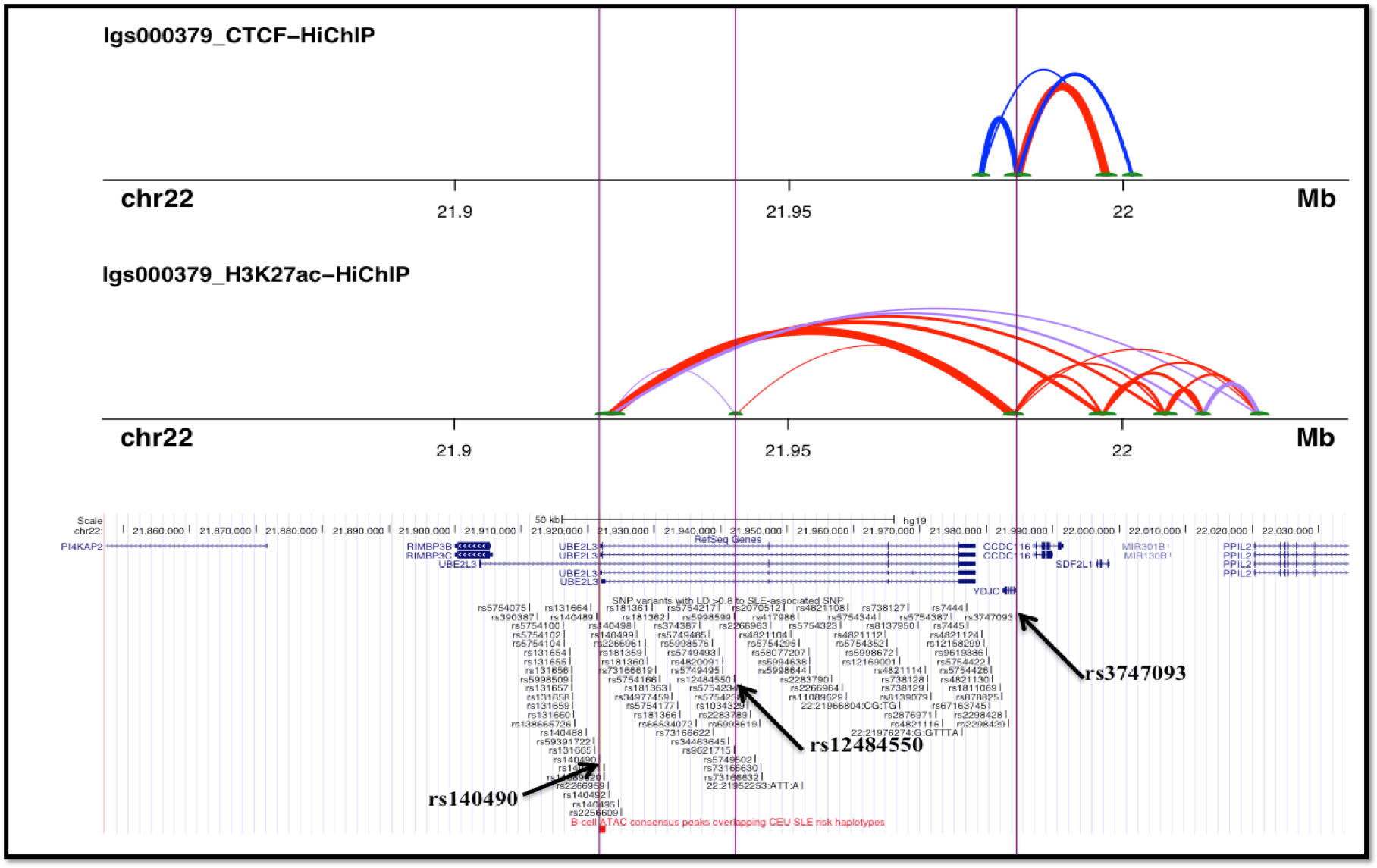
Five variants within the UBE2L3 risk haplotype that overlap the chromatin accessible regions (Assay for Transposase Accessible Chromatin – ATAC peaks) in T or B lymphocytes or Monocytes from SLE patients were selected for screening causal variant candidates. Electrophoretic Mobility Shift Assays (EMSAs) were performed to evaluate the differential binding ability of non-risk and risk alleles of individual variants to nuclear proteins. Nuclear proteins were extracted from T lymphocytes (Jurkat cells) with or without PMA/Ionomycin stimulation for 2hrs. Three-Dimensional (3D) chromatin topology modeling was used to identify physical interactions among the variants. 3D DNA looping data was generated by HiChIP assay to CCCTC-binding factor (CTCF) and histone 3 lysine 27 acetylation (H3K27ac) in EBV-transformed human B cell lines.
In EMSA assays, variants rs140490, rs12484550, and rs3747093 showed increased binding of nuclear protein complexes to the risk alleles compared to non-risk when incubated with Jurkat cell nuclear extracts, with or without stimulation. Variant rs9621715 demonstrated increased binding to the risk allele only in the unstimulated nuclear extract. Variant rs5998599 showed decreased binding to the risk allele compared to the non-risk in nuclear extracts with or without stimulation. Increased binding of nuclear proteins observed in 4 out of the 5 risk variants tested, is suggestive of enhanced transcription factor binding and might therefore account for increased UBE2L3 expression associated with SLE risk.
3D chromatin architecture regulates gene expression by facilitating long-range interaction between distant enhancer and promoter elements in spatial proximity. Our 3D DNA looping data reveals that YdjC chitooligosaccharide deacetylase homolog (YDJC) promoter (rs3747093) interacts with UBE2L3 promoter (rs140490) via H3K27ac loop and with 3’UTR of UBE2L3 via CTCF loop. The risk variants in the first intron (rs12484550, rs9621715, rs5998599) of UBE2L3, also extends H3K27ac loops to YDJC and UBE2L3 promoter elements. 3D chromatin landscape of these risk variants, thus demonstrates significant long-range interactions with the UBE2L3 promoter and could promote UBE2L3 expression.
Our results suggest that SLE risk variant candidates function through enhanced binding of nuclear protein complexes that may likely enhance the strength of long-range DNA looping interactions with UBE2L3 promoter to promote enhanced UBE2L3 expression associated with SLE risk. Genome editing techniques will be used to further explore each of these individual risk variant candidates.